Get a scatter plot showing correlation between replicates.
Source:R/viz_related.R
get_pairwise_corr_plot.RdGet a scatter plot showing correlation between replicates.
Arguments
- x
an abject of class "parcutils". This is an output of the function
run_deseq_analysis().- samples
a character vector denoting samples to plot in scatter plot, default
NULL. If set to NULL all samples are accounted.- genes
a character vector denoting genes to consider in scatter plot, default
NULL. If set to NULL all genes are accounted.#'
Examples
count_file <- system.file("extdata","toy_counts.txt" , package = "parcutils")
count_data <- readr::read_delim(count_file, delim = "\t")
#> Rows: 5000 Columns: 10
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (1): gene_id
#> dbl (9): control_rep1, control_rep2, control_rep3, treat1_rep1, treat1_rep2,...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
sample_info <- count_data %>% colnames() %>% .[-1] %>%
tibble::tibble(samples = . , groups = rep(c("control" ,"treatment1" , "treatment2"), each = 3) )
res <- run_deseq_analysis(counts = count_data ,
sample_info = sample_info,
column_geneid = "gene_id" ,
group_numerator = c("treatment1", "treatment2") ,
group_denominator = c("control"))
#> ℹ Running DESeq2 ...
#> converting counts to integer mode
#> Warning: some variables in design formula are characters, converting to factors
#> estimating size factors
#> estimating dispersions
#> gene-wise dispersion estimates
#> mean-dispersion relationship
#> final dispersion estimates
#> fitting model and testing
#> ✔ Done.
#> ℹ Summarizing DEG ...
#> ✔ Done.
cp <- get_pairwise_corr_plot(res)
names(cp) %>% print()
#> [1] "treatment1" "control" "treatment2"
cp[1] %>% print
#> $treatment1
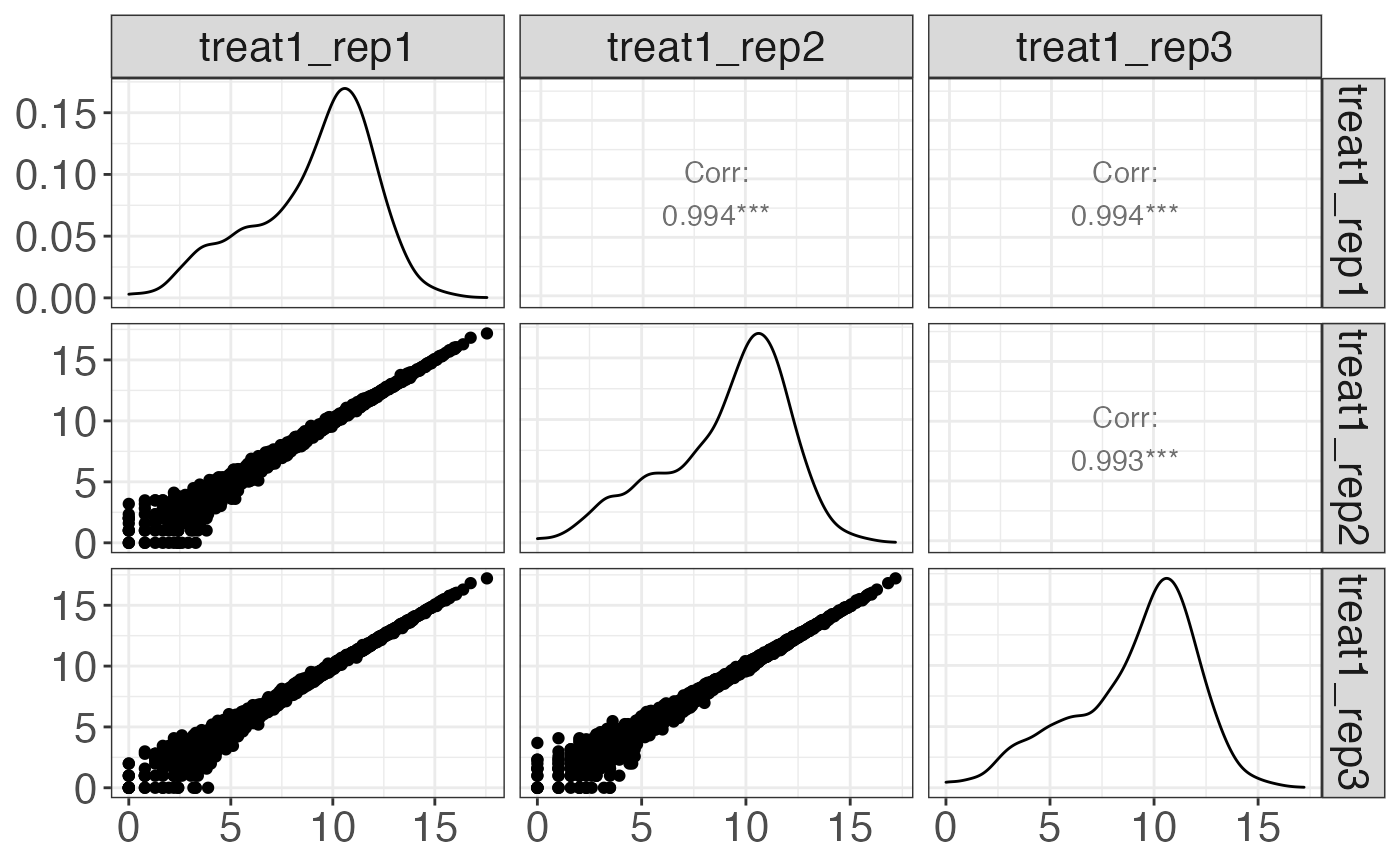 #>
cp[2] %>% print
#> $control
#>
cp[2] %>% print
#> $control
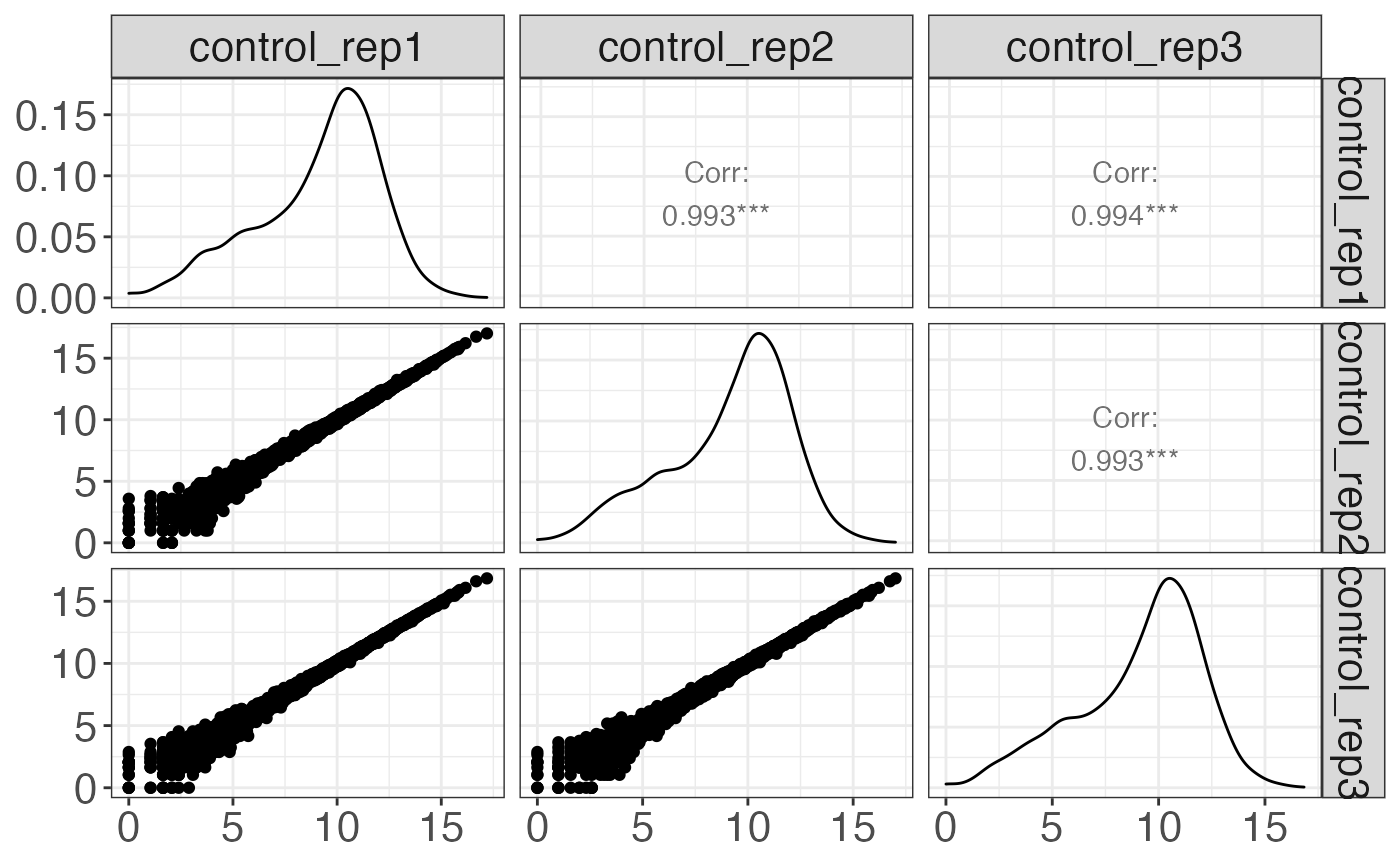 #>
cp[3] %>% print
#> $treatment2
#>
cp[3] %>% print
#> $treatment2
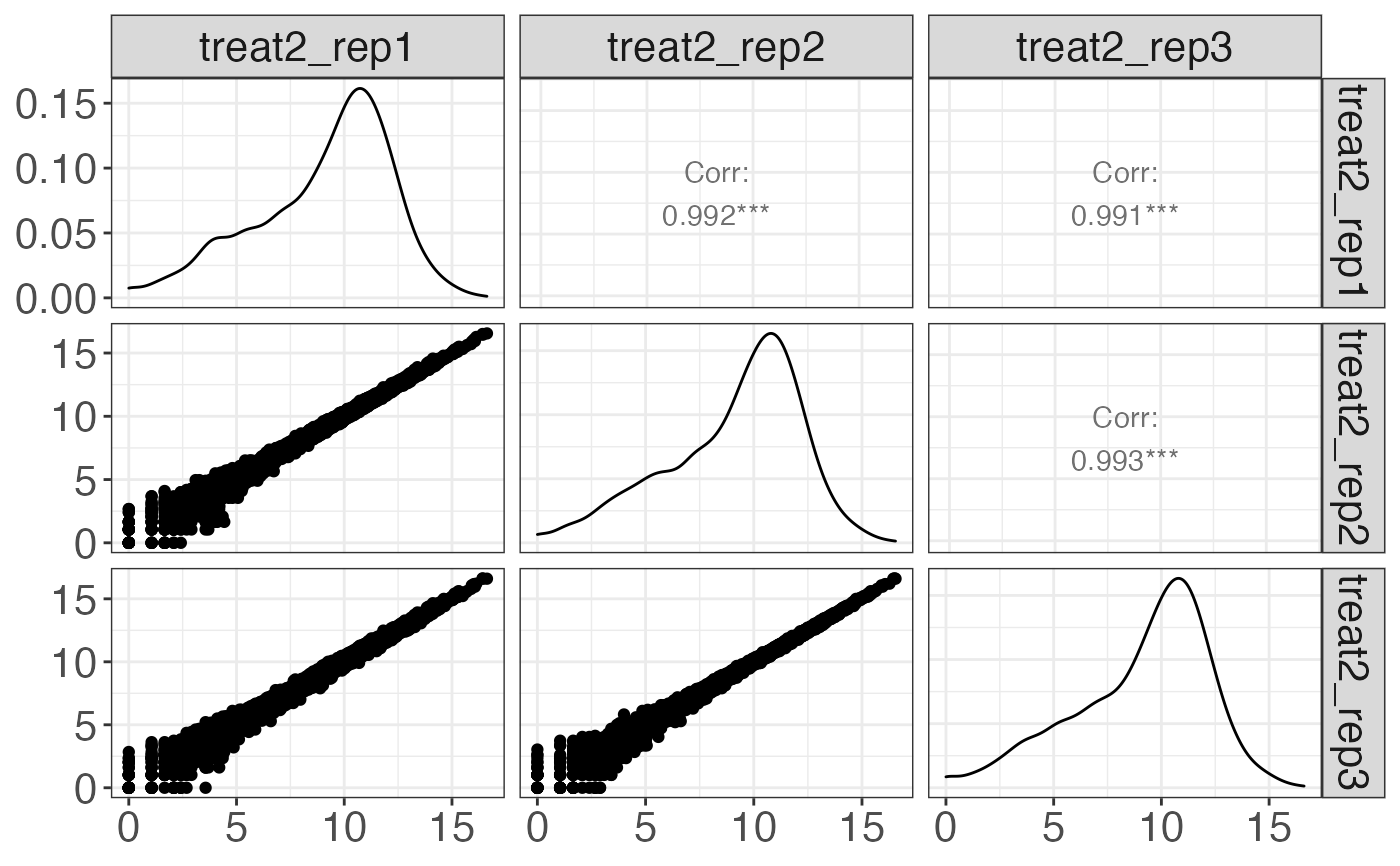 #>
#>