phyloR is an R package to deal with NCBI-BLAST output. It helps to pre-process BLAST tabular output for downstream phylogenetic analysis.
Install
if(require("devtools")){ devtools::install_github("cparsania/phyloR") } else{ install.packages("devtools") devtools::install_github("cparsania/phyloR") }
Assign column names to blast tabular output
blast_out_file <- system.file("extdata" ,"blast_output_01.txt", package = "phyloR") blast_out_tbl <- readr::read_delim(blast_out_file , delim = "\t" , comment = "#" ,col_names = F) colnames(blast_out_tbl) <- phyloR::get_blast_outformat_7_colnames() colnames(blast_out_tbl) #> [1] "query_acc_ver" "subject_acc_ver" "identity" "alignment_length" #> [5] "mismatches" "gap_opens" "q_start" "q_end" #> [9] "s_start" "s_end" "evalue" "bit_score" #> [13] "positives"
Filter blast hits
One can filter blast hits by individual or combinations of these variables.
- e-value
- bitscore
- query coverage
- percent identity
## evalue <= 1e-5 blast_out_tbl %>% phyloR::filter_blast_hits(evalue = 1e-5) #> # A tibble: 545 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 KAE8371401.1 100 253 0 0 #> 2 KAE8371401.1 XP_031924425.1 71.3 247 71 0 #> 3 KAE8371401.1 XP_031940570.1 66.4 247 83 0 #> 4 KAE8371401.1 KAB8261773.1 66.0 247 84 0 #> 5 KAE8371401.1 XP_022392557.1 64.4 247 69 1 #> 6 KAE8371401.1 OQD81666.1 61.7 248 95 0 #> 7 KAE8371401.1 GES63448.1 39.3 229 135 3 #> 8 KAE8371401.1 XP_001214927.1 39.3 229 135 3 #> 9 KAE8371401.1 OQD86157.1 38.6 228 132 3 #> 10 KAE8371401.1 XP_002375911.1 39.1 230 129 5 #> # … with 535 more rows, and 7 more variables: q_start <dbl>, q_end <dbl>, #> # s_start <dbl>, s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl> ## Filter blast hits : bitscore >= 200 blast_out_tbl %>% phyloR::filter_blast_hits(bit_score = 200) #> # A tibble: 6 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 KAE8371401.1 100 253 0 0 #> 2 KAE8371401.1 XP_031924425.1 71.3 247 71 0 #> 3 KAE8371401.1 XP_031940570.1 66.4 247 83 0 #> 4 KAE8371401.1 KAB8261773.1 66.0 247 84 0 #> 5 KAE8371401.1 XP_022392557.1 64.4 247 69 1 #> 6 KAE8371401.1 OQD81666.1 61.7 248 95 0 #> # … with 7 more variables: q_start <dbl>, q_end <dbl>, s_start <dbl>, #> # s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl> ## Filter blast hits : query coverage >= 90 blast_out_tbl %>% phyloR::filter_blast_hits(query_cov = 90, query_length = 253) #> # A tibble: 89 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 KAE8371401.1 100 253 0 0 #> 2 KAE8371401.1 XP_031924425.1 71.3 247 71 0 #> 3 KAE8371401.1 XP_031940570.1 66.4 247 83 0 #> 4 KAE8371401.1 KAB8261773.1 66.0 247 84 0 #> 5 KAE8371401.1 XP_022392557.1 64.4 247 69 1 #> 6 KAE8371401.1 OQD81666.1 61.7 248 95 0 #> 7 KAE8371401.1 GES63448.1 39.3 229 135 3 #> 8 KAE8371401.1 XP_001214927.1 39.3 229 135 3 #> 9 KAE8371401.1 XP_002375911.1 39.1 230 129 5 #> 10 KAE8371401.1 OQD72420.1 40 230 127 5 #> # … with 79 more rows, and 7 more variables: q_start <dbl>, q_end <dbl>, #> # s_start <dbl>, s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl> ## Filter blast hits : percent identity >= 90 blast_out_tbl %>% phyloR::filter_blast_hits(identity = 90) #> # A tibble: 1 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 KAE8371401.1 100 253 0 0 #> # … with 7 more variables: q_start <dbl>, q_end <dbl>, s_start <dbl>, #> # s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl> ## Filter blast hits : evalue <= 1e-5, bitscore >= 200, query coverage >= 90, percent identity >= 60, blast_out_tbl %>% phyloR::filter_blast_hits(evalue = 1e-5, bit_score = 200, query_cov = 90, query_length = 253, identity = 60) #> # A tibble: 6 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 KAE8371401.1 100 253 0 0 #> 2 KAE8371401.1 XP_031924425.1 71.3 247 71 0 #> 3 KAE8371401.1 XP_031940570.1 66.4 247 83 0 #> 4 KAE8371401.1 KAB8261773.1 66.0 247 84 0 #> 5 KAE8371401.1 XP_022392557.1 64.4 247 69 1 #> 6 KAE8371401.1 OQD81666.1 61.7 248 95 0 #> # … with 7 more variables: q_start <dbl>, q_end <dbl>, s_start <dbl>, #> # s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl>
Format fasta headers
Fasta file downloaded as an output of blast result has fasta headers of two types
1) Headers with alignment co-ordinates - if only aligned sequences downloaded.
- E.g.
KAE8371401.1:1-253 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus bertholletius]
2) Headers without alignment co-ordinates - if full sequences downloaded.
- E.g.
KAE8371401.1 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus bertholletius]
By default, phyloR::format_fasta_headers() divides headers in three (header type 2) or four (header type 1) groups delimited by 2 underscores (__).
1) subject id 2) alignment coordinates (Optional for header type 1) 3) subject description 4) subject species
One can drop alignment coordinates from sequence headers by keeping argument keep_alignemnt_coord = FALSE
fa_file <- system.file("extdata" ,"blast_output_01.fasta", package = "phyloR") fa <- Biostrings::readBStringSet(fa_file) ## existing headers names(fa) %>% head() #> [1] "KAE8371401.1:1-253 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus bertholletius]" #> [2] "XP_031924425.1:1-247 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus caelatus]" #> [3] "XP_031940570.1:1-247 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus pseudonomius]" #> [4] "KAB8261773.1:1-247 trypsin-like cysteine/serine peptidase domain-containing protein [Aspergillus pseudonomius]" #> [5] "XP_022392557.1:1-228 hypothetical protein ABOM_003016 [Aspergillus bombycis]" #> [6] "OQD81666.1:1-248 hypothetical protein PENANT_c026G03216 [Penicillium antarcticum]" ## new headers fa_file %>% phyloR::format_fasta_headers() %>% names() %>% head() #> [1] "KAE8371401_1__1_253__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_bertholletius" #> [2] "XP_031924425_1__1_247__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_caelatus" #> [3] "XP_031940570_1__1_247__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_pseudonomius" #> [4] "KAB8261773_1__1_247__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_pseudonomius" #> [5] "XP_022392557_1__1_228__hypothetical_protein_ABOM_003016__Aspergillus_bombycis" #> [6] "OQD81666_1__1_248__hypothetical_protein_PENANT_c026G03216__Penicillium_antarcticum" ## drop alignment co-ordinates fa_file %>% phyloR::format_fasta_headers(keep_alignemnt_coord = FALSE) %>% names() %>% head() #> [1] "KAE8371401_1__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_bertholletius" #> [2] "XP_031924425_1__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_caelatus" #> [3] "XP_031940570_1__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_pseudonomius" #> [4] "KAB8261773_1__trypsin_like_cysteine_serine_peptidase_domain_containing_protein__Aspergillus_pseudonomius" #> [5] "XP_022392557_1__hypothetical_protein_ABOM_003016__Aspergillus_bombycis" #> [6] "OQD81666_1__hypothetical_protein_PENANT_c026G03216__Penicillium_antarcticum"
Remove redundant hits from blast tabular ouput
BLAST reports same subject hit multiple time with different alignment co-ordinates. For a same subjet id, phyloR::remove_redundant_hits() select longest subject hit out of given multiples.
blast_out_tbl %>% phyloR::remove_redundant_hits() #> # A tibble: 716 x 13 #> query_acc_ver subject_acc_ver identity alignment_length mismatches gap_opens #> <chr> <chr> <dbl> <dbl> <dbl> <dbl> #> 1 KAE8371401.1 1FN8_A 31.6 209 127 6 #> 2 KAE8371401.1 1GDU_A 31.9 210 127 6 #> 3 KAE8371401.1 1PPZ_A 31.9 210 127 6 #> 4 KAE8371401.1 AAQ04074.1 27.5 218 135 9 #> 5 KAE8371401.1 AAR91718.1 30.8 117 73 2 #> 6 KAE8371401.1 AAR91719.1 28.7 157 94 6 #> 7 KAE8371401.1 AAW31593.1 27.2 228 147 6 #> 8 KAE8371401.1 ADK37838.1 29.5 227 136 9 #> 9 KAE8371401.1 ADY16698.1 35.5 242 151 4 #> 10 KAE8371401.1 AER28315.1 30.5 236 148 5 #> # … with 706 more rows, and 7 more variables: q_start <dbl>, q_end <dbl>, #> # s_start <dbl>, s_end <dbl>, evalue <dbl>, bit_score <dbl>, positives <dbl>
Subset fasta sequences
After filtering hits from tabular blast output, next obvious question is to filter same sequences from a fasta file and write them to new file. phyloR::subset_bstringset() subset the sequences from a fasta using a vector of fasta headers.
fa_file <- system.file("extdata" ,"blast_output_01.fasta", package = "phyloR") fa <- Biostrings::readBStringSet(fa_file) query_headers <- fa %>% names() %>% sample(100) seq_filtered <- phyloR::subset_bstringset(x = query_headers , y = fa, partial_match = F) # One can use function `Biostrings::writeXStringSet()` to write filterd sequences to new fasta file.
Assign taxonomy columns to NCBI protein acession
phyloR allows you to assign NCBI taxonomy levels to NCBI protein accession using function phyloR::add_taxonomy_columns(). One of the applications of this function is to assign the NCBI taxonomy levels to blast output, which is required for downstream phylogenetic analysis. For example, a phylogenetic tree generated from blast hits often required to color resultant tree branches either by kingdom, family or any other NCBI taxonomy level. One can easily identify these taxonomy levels using function phyloR::add_taxonomy_columns()
## add kingdom with_kingdom <- blast_out_tbl %>% slice(1:10) %>% phyloR::add_taxonomy_columns(ncbi_accession_colname = "subject_acc_ver", taxonomy_level = "kingdom") #> No ENTREZ API key provided #> Get one via taxize::use_entrez() #> See https://ncbiinsights.ncbi.nlm.nih.gov/2017/11/02/new-api-keys-for-the-e-utilities/ #> ✓ Done. Time taken 5.23 #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.08 with_kingdom %>% dplyr::select(query_acc_ver, subject_acc_ver, kingdom) #> # A tibble: 10 x 3 #> query_acc_ver subject_acc_ver kingdom #> <chr> <chr> <chr> #> 1 KAE8371401.1 KAE8371401.1 Fungi #> 2 KAE8371401.1 XP_031924425.1 Fungi #> 3 KAE8371401.1 XP_031940570.1 Fungi #> 4 KAE8371401.1 KAB8261773.1 Fungi #> 5 KAE8371401.1 XP_022392557.1 Fungi #> 6 KAE8371401.1 OQD81666.1 Fungi #> 7 KAE8371401.1 GES63448.1 Fungi #> 8 KAE8371401.1 XP_001214927.1 Fungi #> 9 KAE8371401.1 OQD86157.1 Fungi #> 10 KAE8371401.1 XP_002375911.1 Fungi ## add family with_kingdom_and_family <- with_kingdom %>% phyloR::add_taxonomy_columns(ncbi_accession_colname = "subject_acc_ver" , taxonomy_level = "family") #> ── WARNING ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ℹ As column 'taxid' present in blast output tbl, same will be used to map taxonomy level. #> ℹ To perform new 'taxid' search either remove or rename columnn 'taxid'. #> ── WARNING ENDS ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.05 with_kingdom_and_family %>% dplyr::select(query_acc_ver, subject_acc_ver, kingdom, family ) #> # A tibble: 10 x 4 #> query_acc_ver subject_acc_ver kingdom family #> <chr> <chr> <chr> <chr> #> 1 KAE8371401.1 KAE8371401.1 Fungi Aspergillaceae #> 2 KAE8371401.1 XP_031924425.1 Fungi Aspergillaceae #> 3 KAE8371401.1 XP_031940570.1 Fungi Aspergillaceae #> 4 KAE8371401.1 KAB8261773.1 Fungi Aspergillaceae #> 5 KAE8371401.1 XP_022392557.1 Fungi Aspergillaceae #> 6 KAE8371401.1 OQD81666.1 Fungi Aspergillaceae #> 7 KAE8371401.1 GES63448.1 Fungi Aspergillaceae #> 8 KAE8371401.1 XP_001214927.1 Fungi Aspergillaceae #> 9 KAE8371401.1 OQD86157.1 Fungi Aspergillaceae #> 10 KAE8371401.1 XP_002375911.1 Fungi Aspergillaceae ## add species with_kingdom_family_and_species <- with_kingdom_and_family %>% phyloR::add_taxonomy_columns(ncbi_accession_colname = "subject_acc_ver" , taxonomy_level = "species") #> ── WARNING ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ℹ As column 'taxid' present in blast output tbl, same will be used to map taxonomy level. #> ℹ To perform new 'taxid' search either remove or rename columnn 'taxid'. #> ── WARNING ENDS ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.04 with_kingdom_family_and_species %>% dplyr::select(query_acc_ver, subject_acc_ver, kingdom, family ,species) #> # A tibble: 10 x 5 #> query_acc_ver subject_acc_ver kingdom family species #> <chr> <chr> <chr> <chr> <chr> #> 1 KAE8371401.1 KAE8371401.1 Fungi Aspergillaceae Aspergillus bertholleti… #> 2 KAE8371401.1 XP_031924425.1 Fungi Aspergillaceae Aspergillus caelatus #> 3 KAE8371401.1 XP_031940570.1 Fungi Aspergillaceae Aspergillus pseudonomius #> 4 KAE8371401.1 KAB8261773.1 Fungi Aspergillaceae Aspergillus pseudonomius #> 5 KAE8371401.1 XP_022392557.1 Fungi Aspergillaceae Aspergillus bombycis #> 6 KAE8371401.1 OQD81666.1 Fungi Aspergillaceae Penicillium antarcticum #> 7 KAE8371401.1 GES63448.1 Fungi Aspergillaceae Aspergillus terreus #> 8 KAE8371401.1 XP_001214927.1 Fungi Aspergillaceae Aspergillus terreus #> 9 KAE8371401.1 OQD86157.1 Fungi Aspergillaceae Penicillium antarcticum #> 10 KAE8371401.1 XP_002375911.1 Fungi Aspergillaceae Aspergillus flavus
Use NCBI taxonomy levels to color the phyloegenetic tree
An object of class phylo is widely used object to visualize the phylogenetic tree in R. To gain more insights from the resultant phylogenetic tree, often tree tips required to color by different levels of taxonomy, for instance species, kingdom or family. Obtaining taxonomy data to use them for tree annotations is not straight forward task and require lots of data wrangling in R. phyloR provides handy way to map such a taxonomy data to the phylogenetic tree. For example, one can convert an object of class phylo to an object of class tibble and vice versa. Once the tibble obtained, phyloR allows to add columns of ncbi taxonomy levels. Resultant tibble can be passed to ggtree::ggtree() to visualize the tree and taxonomy levels can be used, for instance, to color the tree tips.
tree_string <- "((XP_005187699_1__Musca_domestica:0.070627277,(XP_019893806_1__Musca_domestica:0.071069674,((XP_013113221_1__Stomoxys_calcitrans:0.1494662042,ACB98719_1__Glossina_morsitans_morsitans:0.3489851076)67.4/100:0.0470213767,XP_013102958_1__Stomoxys_calcitrans:0.1794878827)98.1/100:0.0959227604)88.2/99:0.0323598861)93/99:0.0435291148,((XP_017472861_1__Rhagoletis_zephyria:0.0049337059,XP_017472862_1__Rhagoletis_zephyria:0.0112391294)97.3/100:0.0860969479,(XP_020713236_1__Ceratitis_capitata:0.2642805176,(XP_014102010_1__Bactrocera_oleae:0.1183517872,XP_018784523_1__Bactrocera_latifrons:0.1137567198)29.6/88:0.0758551876)99.9/100:0.247740081)92/100:0.0716529011)34.3/66:2.487103817;" tree_objct <- treeio::read.tree(text = tree_string) tree_tbl <- tree_objct %>% ggtree::fortify() tree_tbl <- tree_tbl %>% dplyr::mutate( seqid = dplyr::case_when(isTip ~ stringr::str_replace(label , pattern = "__.*","" ) %>% ## split by '__' stringr::str_replace(pattern = "_\\d$" , ""), ## remove trailing digits from seqid TRUE ~ label ) ) ## add taxonomy tree_tbl_with_taxonomy <- tree_tbl %>% phyloR::tidy_taxonomy_tree(ncbi_accession_colname = "seqid",taxonomy_levels = c("species" ,"kingdom","family")) #> ✓ Done. Time taken 3.3 #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.05 #> ── WARNING ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ── WARNING ENDS ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.04 #> ── WARNING ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ── WARNING ENDS ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ● Rank search begins... #> ────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── #> ✓ Done. Time taken 0.07 ## visualize tree # tips colored by species tree_tbl_with_taxonomy %>% ggtree::ggtree() + ggtree::geom_tiplab(ggplot2::aes(color = species, label = seqid),size = 20) + ggplot2::theme(legend.position = "bottom" , legend.text = ggplot2::element_text(size= 50) , legend.title = ggplot2::element_text(size= 50))
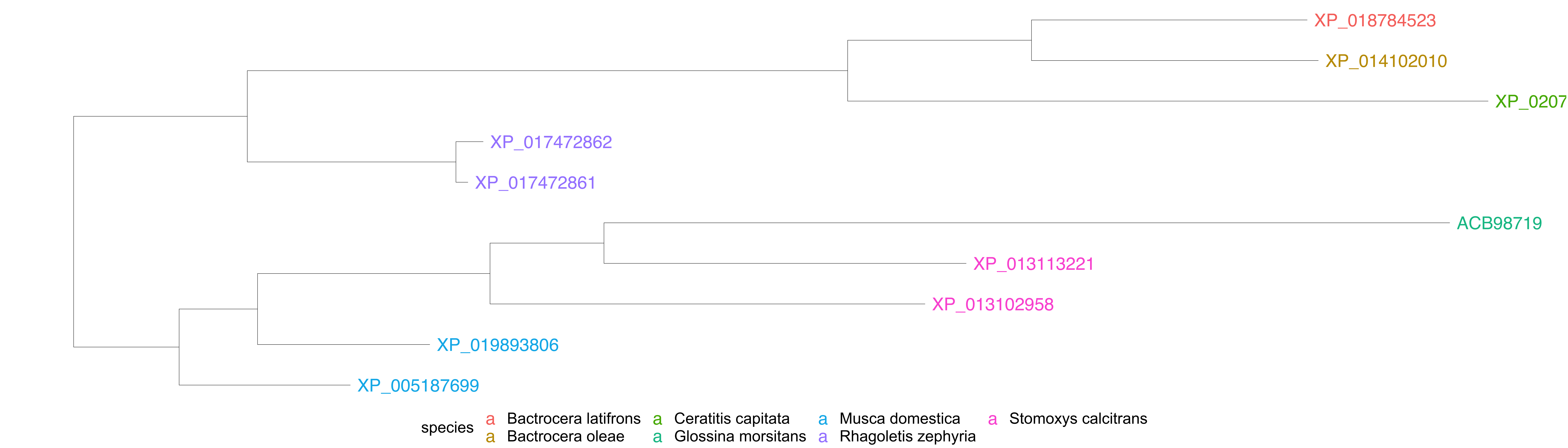
# tips colored by family tree_tbl_with_taxonomy %>% ggtree::ggtree() + ggtree::geom_tiplab(ggplot2::aes(color = family,label = seqid),size = 20) + ggplot2::theme(legend.position = "bottom" , legend.text = ggplot2::element_text(size= 50) , legend.title = ggplot2::element_text(size= 50))

# tips colored by kingdom tree_tbl_with_taxonomy %>% ggtree::ggtree() + ggtree::geom_tiplab(ggplot2::aes(color = kingdom,label = seqid ),size =20) + ggplot2::theme(legend.position = "bottom" , legend.text = ggplot2::element_text(size= 50) , legend.title = ggplot2::element_text(size= 50))
