Deterministic Multithreaded Genomic Interval Operations
![]()
![]()
fastRanges is a multithreaded interval engine for IRanges and GRanges. It keeps Bioconductor-style overlap semantics and familiar argument grammar while targeting the workloads that usually dominate runtime in genomics: large findOverlaps() jobs, repeated query batches against one subject, and overlap-derived summaries such as counts, joins, and aggregation.
Website: https://cparsania.github.io/fastRanges/
Source: https://github.com/cparsania/fastRanges
Installation
Bioconductor
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("fastRanges")GitHub
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
remotes::install_github("cparsania/fastRanges", ref = "main")Quick Start
library(fastRanges)
library(GenomicRanges)
data("fast_ranges_example", package = "fastRanges")
query <- fast_ranges_example$query
subject <- fast_ranges_example$subject
# One-off overlap call
hits <- fast_find_overlaps(query, subject, threads = 4)
# Repeated-query workflow
subject_index <- fast_build_index(subject)
hits_indexed <- fast_find_overlaps(query, subject_index, threads = 4)
# Derived summaries
counts <- fast_count_overlaps(query, subject_index, threads = 4)
joined <- fast_overlap_join(query, subject, threads = 4)The package ships a small in-memory example object and matching BED files:
data("fast_ranges_example", package = "fastRanges")
names(fast_ranges_example)
system.file("extdata", "query_peaks.bed", package = "fastRanges")
system.file("extdata", "subject_genes.bed", package = "fastRanges")Function Grammar
Overlap Grammar
-
fast_find_overlaps(): return overlap pairs asHits -
fast_count_overlaps(): per-query overlap counts -
fast_overlaps_any(): per-query logical overlap flag -
fast_build_index(): build a reusable subject index
Join Grammar
-
fast_overlap_join(): overlap join withjoin = "inner"or"left" -
fast_inner_overlap_join(),fast_left_overlap_join() -
fast_semi_overlap_join(),fast_anti_overlap_join()
Compatibility
fastRanges is designed to stay close to Bioconductor overlap semantics for supported inputs, but it is currently best viewed as a high-throughput engine for IRanges and GRanges, not as a blanket replacement for every findOverlaps() input class.
Currently supported:
IRangesGRanges-
select = "all","first","last", and"arbitrary" - empty-range handling with Bioconductor-compatible fallback behavior
Currently unsupported:
- circular genomic sequences
GRangesList
Unsupported inputs are rejected explicitly with a clear error.
Benchmark Highlights
Saved benchmark results on a 96-core Linux server show:
- about
5.19xto5.40xGRangesspeedup for indexedfastRangesversusGenomicRanges::findOverlaps() - about
4.90xspeedup in repeated-query workloads when the subject index is reused - continued scaling on dense
GRangesand largeIRangesworkloads - retained gains in grouped counting and overlap aggregation
| GRanges speedup vs baseline | Repeated-query speedup |
|---|---|
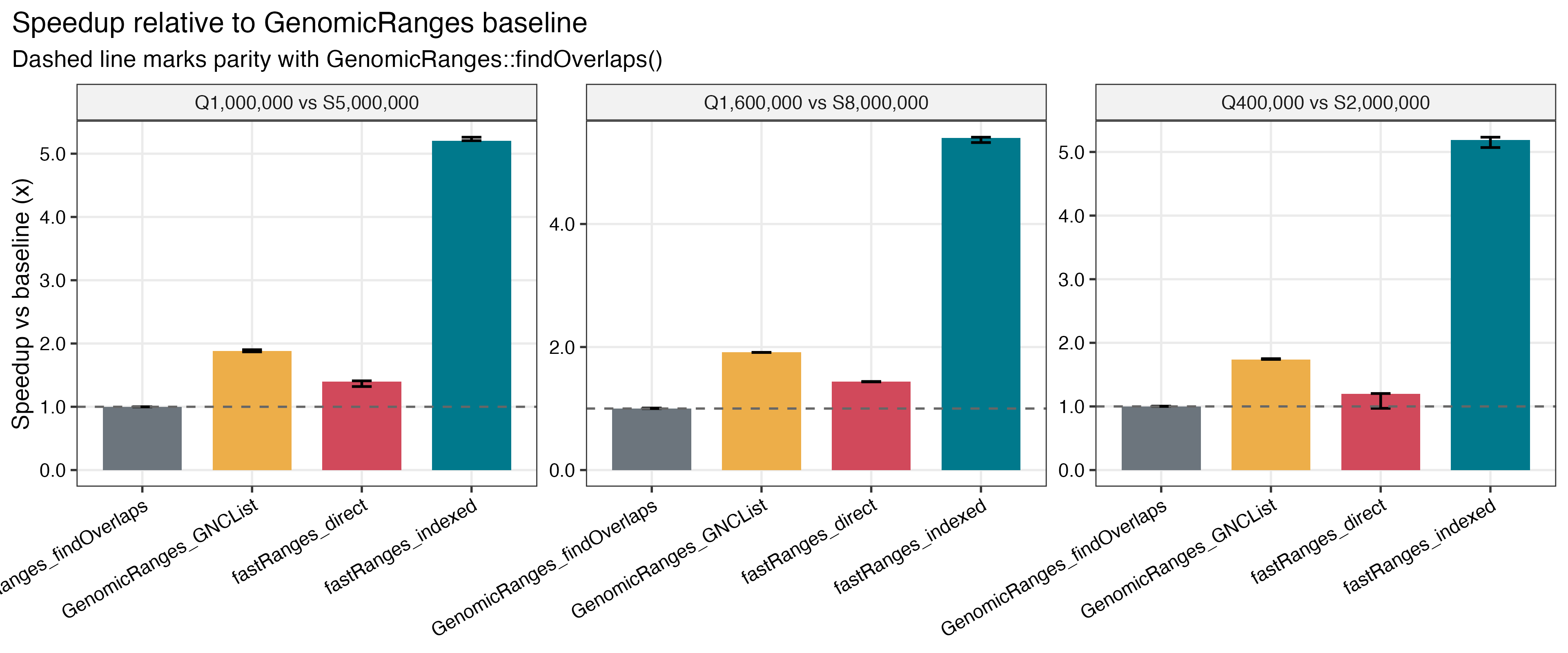 |
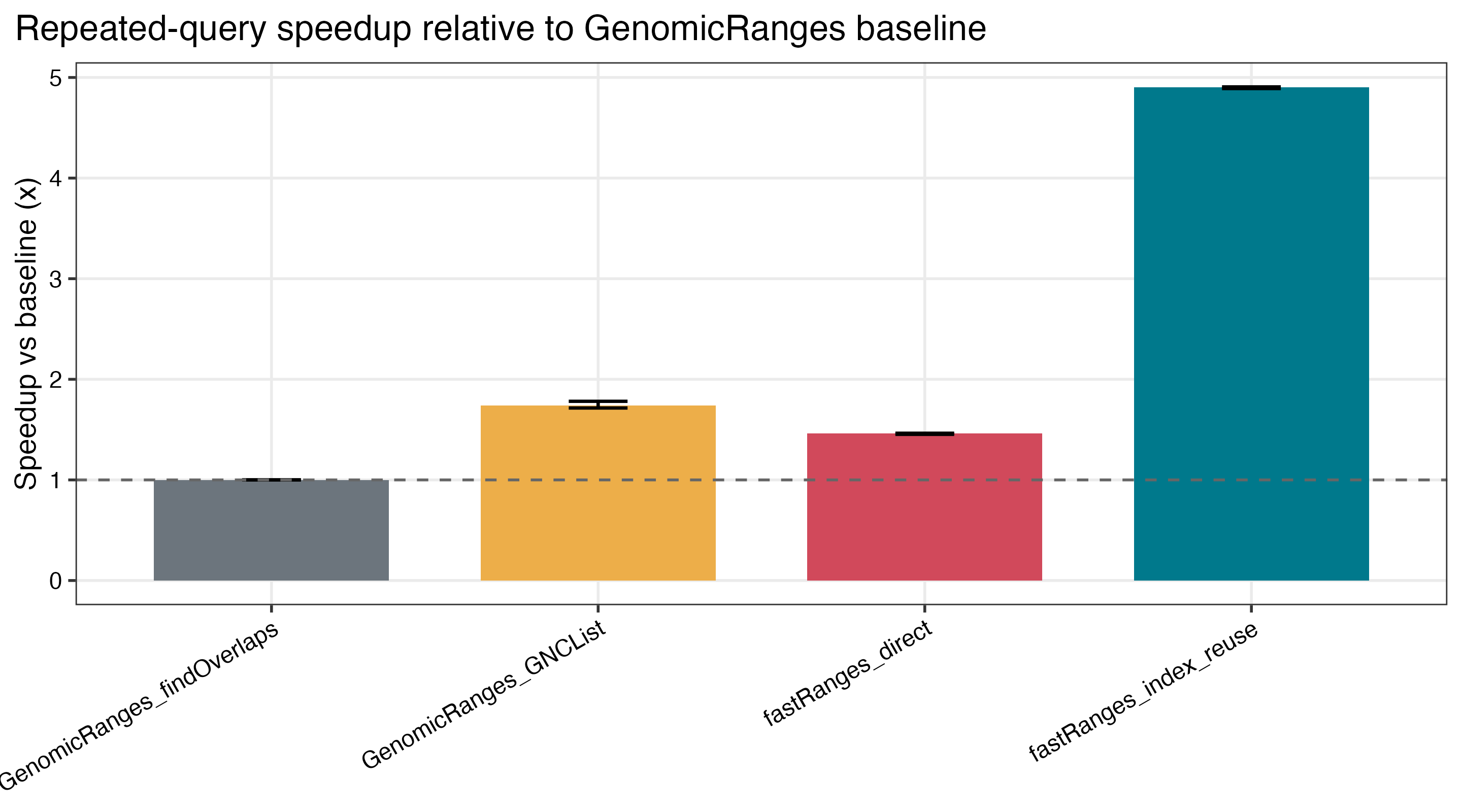 |
| Dense GRanges scaling | IRanges absolute runtime |
|---|---|
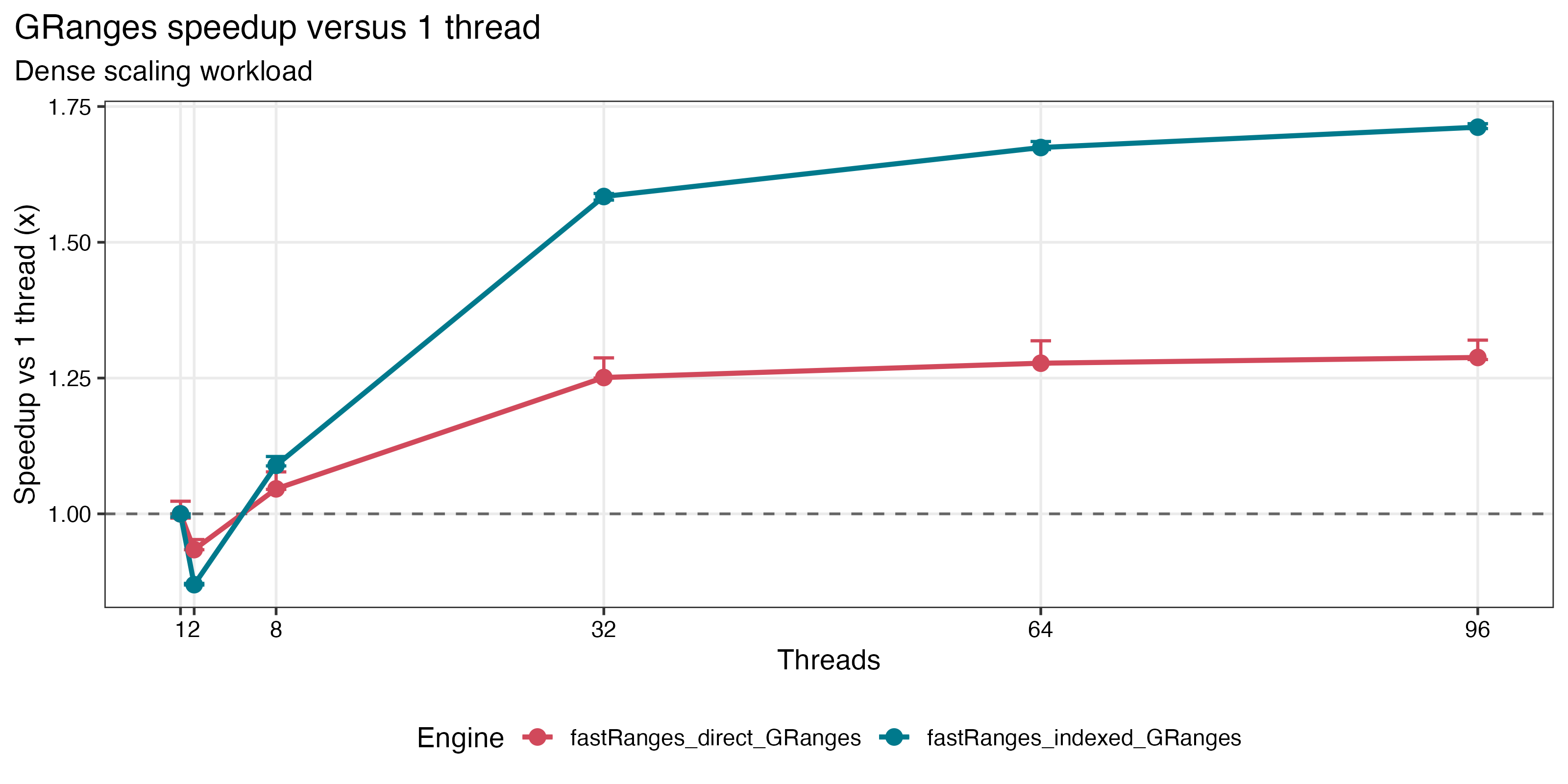 |
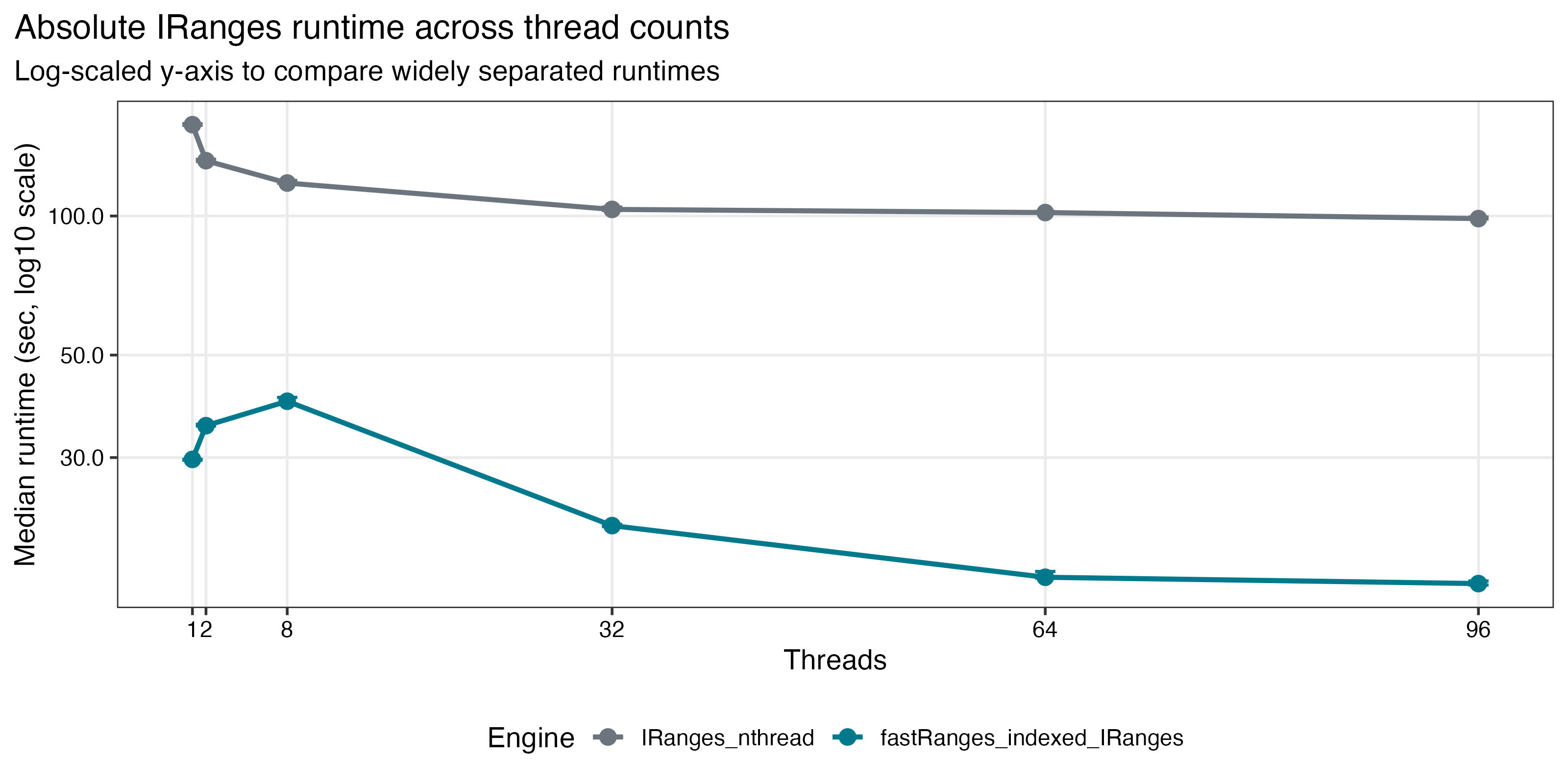 |
Benchmark resources:
Practical Use
- Use direct mode for one-off overlap calls.
- Use
fast_build_index(subject)when the same annotation is queried many times. - Use higher
threadsfor large workloads on multicore machines. - Keep
deterministic = TRUEwhen stable output ordering matters. - Use
deterministic = FALSEwhen maximum multithreaded throughput matters more than stable hit ordering.