Cell-Type Deconvolution with VISTA (airway)
VISTA Development Team
VISTA-deconvolution.RmdOverview
This vignette demonstrates how to run cell-type
deconvolution with VISTA using the Bioconductor
airway dataset.
Workflow covered:
- Build a
VISTAobject from airway counts and metadata. - Run
run_cell_deconvolution()(xCell2 backend). - Extract cell-fraction estimates with
get_cell_fractions(). - Visualize sample-level composition with
get_celltype_barplot(). - Compare treatment groups with
get_celltype_group_dotplot(). - Inspect cell-type/sample structure with
get_celltype_heatmap().
Prepare Airway Data
data("airway", package = "airway")
counts_matrix <- SummarizedExperiment::assay(airway, "counts")
sample_metadata <- as.data.frame(SummarizedExperiment::colData(airway))
# Keep sample IDs explicit for VISTA alignment.
sample_metadata$sample_names <- rownames(sample_metadata)
sample_metadata$cond_long <- ifelse(sample_metadata$dex == "trt", "dexamethasone", "untreated")
count_data <- as.data.frame(counts_matrix) %>%
tibble::rownames_to_column("gene_id")
# Ensure column order follows sample_metadata.
count_data <- count_data[, c("gene_id", sample_metadata$sample_names)]
dim(count_data)
#> [1] 63677 9
sample_metadata[, c("sample_names", "cell", "dex", "cond_long")]
#> sample_names cell dex cond_long
#> SRR1039508 SRR1039508 N61311 untrt untreated
#> SRR1039509 SRR1039509 N61311 trt dexamethasone
#> SRR1039512 SRR1039512 N052611 untrt untreated
#> SRR1039513 SRR1039513 N052611 trt dexamethasone
#> SRR1039516 SRR1039516 N080611 untrt untreated
#> SRR1039517 SRR1039517 N080611 trt dexamethasone
#> SRR1039520 SRR1039520 N061011 untrt untreated
#> SRR1039521 SRR1039521 N061011 trt dexamethasoneCreate VISTA Object
vista_airway <- create_vista(
counts = count_data,
sample_info = sample_metadata,
column_geneid = "gene_id",
group_column = "cond_long",
group_numerator = "dexamethasone",
group_denominator = "untreated",
covariates = "cell",
method = "deseq2",
min_counts = 10,
min_replicates = 1
)
# Add gene annotations (used for downstream interpretation and fallback symbol mapping).
vista_airway <- set_rowdata(
vista_airway,
orgdb = org.Hs.eg.db,
columns = c("SYMBOL", "GENENAME", "ENTREZID"),
keytype = "ENSEMBL"
)
vista_airway
#> class: VISTA
#> dim: 18086 8
#> metadata(12): de_results de_summary ... design comparison
#> assays(1): norm_counts
#> rownames(18086): ENSG00000000003 ENSG00000000419 ... ENSG00000273487
#> ENSG00000273488
#> rowData names(4): baseMean SYMBOL GENENAME ENTREZID
#> colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
#> colData names(12): SampleName cell ... sizeFactor sample_namesRun Deconvolution
run_cell_deconvolution() currently uses xCell2.
If xCell2 is unavailable, this vignette will skip
deconvolution sections.
cat("Package 'xCell2' is not installed; deconvolution steps are skipped.\n")
cat("Install it to run these sections:\n")
cat(" Install package 'xCell2' from Bioconductor.\n")
# First try default threshold, then relax minSharedGenes if needed.
deconv_try <- tryCatch(
run_cell_deconvolution(
x = vista_airway,
method = "xCell2",
gene_id_type = "ensembl"
),
error = function(e) e
)
if (inherits(deconv_try, "error")) {
msg <- conditionMessage(deconv_try)
if (grepl("minSharedGenes", msg, fixed = TRUE)) {
message("Retrying xCell2 deconvolution with minSharedGenes = 0.6")
deconv_try <- tryCatch(
run_cell_deconvolution(
x = vista_airway,
method = "xCell2",
gene_id_type = "ensembl",
minSharedGenes = 0.6
),
error = function(e) e
)
}
}
if (inherits(deconv_try, "error")) {
has_deconv <- FALSE
message("Deconvolution could not be completed in this environment:\n", conditionMessage(deconv_try))
} else {
vista_deconv <- deconv_try
has_deconv <- TRUE
vista_deconv
}
#> class: VISTA
#> dim: 18086 8
#> metadata(13): de_results de_summary ... comparison cell_fractions
#> assays(1): norm_counts
#> rownames(18086): ENSG00000000003 ENSG00000000419 ... ENSG00000273487
#> ENSG00000273488
#> rowData names(4): baseMean SYMBOL GENENAME ENTREZID
#> colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
#> colData names(12): SampleName cell ... sizeFactor sample_namesInspect Cell Fractions
cell_fractions <- get_cell_fractions(vista_deconv)
dim(cell_fractions)
#> [1] 8 43
cell_fractions[1:min(4, nrow(cell_fractions)), 1:min(6, ncol(cell_fractions))]
#> neutrophil monocyte megakaryocyte-erythroid progenitor cell
#> SRR1039508 2.746005e-22 0.000000e+00 0.000000e+00
#> SRR1039509 4.648547e-23 0.000000e+00 1.157681e-05
#> SRR1039512 0.000000e+00 1.220985e-20 5.108035e-05
#> SRR1039513 4.611114e-22 0.000000e+00 1.347784e-05
#> CD4-positive, alpha-beta T cell regulatory T cell
#> SRR1039508 2.783925e-05 1.816553e-24
#> SRR1039509 3.985213e-05 0.000000e+00
#> SRR1039512 1.816519e-05 0.000000e+00
#> SRR1039513 2.921939e-05 0.000000e+00
#> central memory CD4-positive, alpha-beta T cell
#> SRR1039508 0.000000e+00
#> SRR1039509 6.472498e-31
#> SRR1039512 2.746680e-23
#> SRR1039513 0.000000e+00Plot Cell-Type Composition
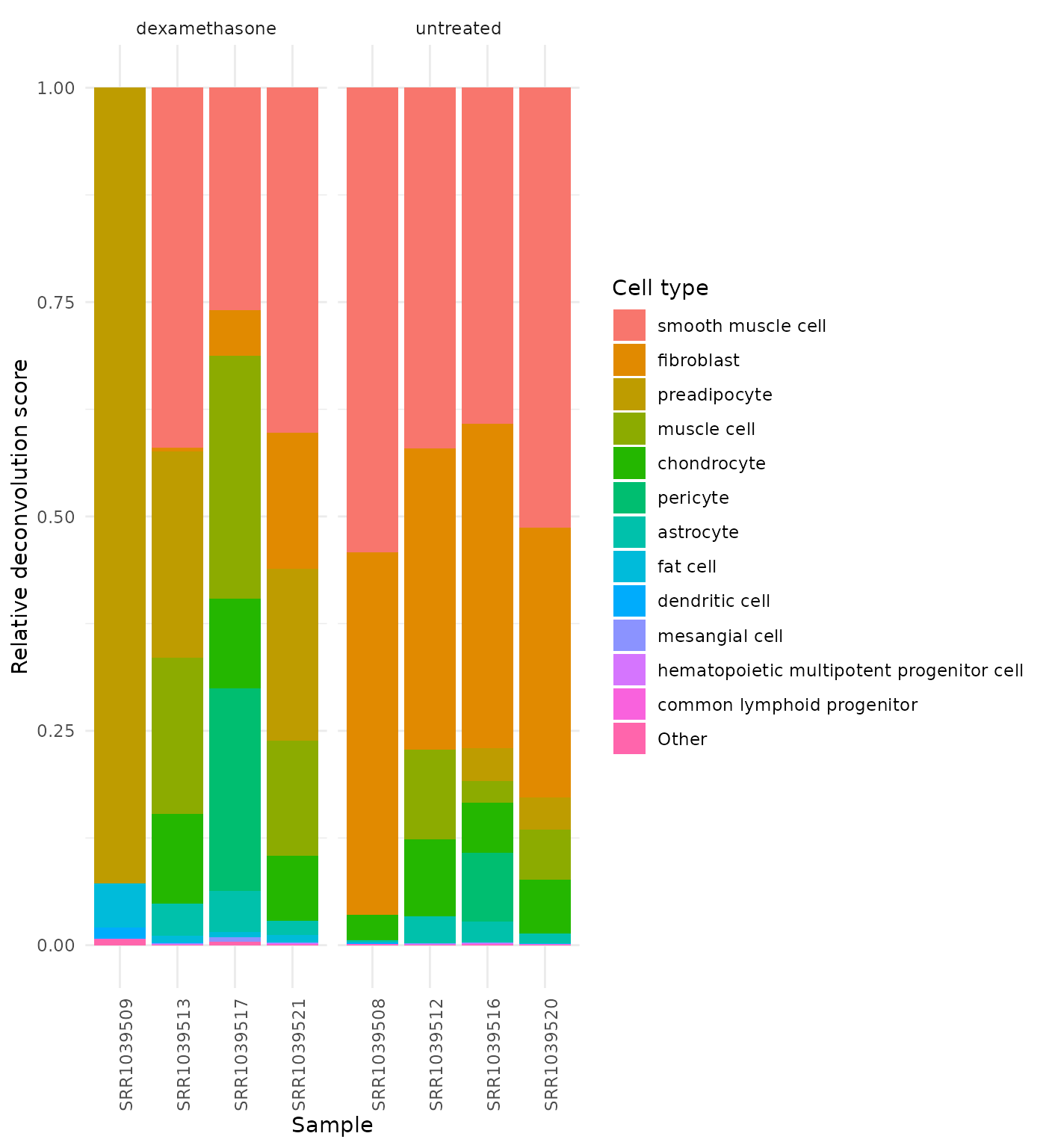
get_celltype_barplot(
x = vista_deconv,
group_column = "cond_long",
top_n = 12,
collapse_other = TRUE,
normalize = "sample",
facet_by = "group",
base_size = 11
)
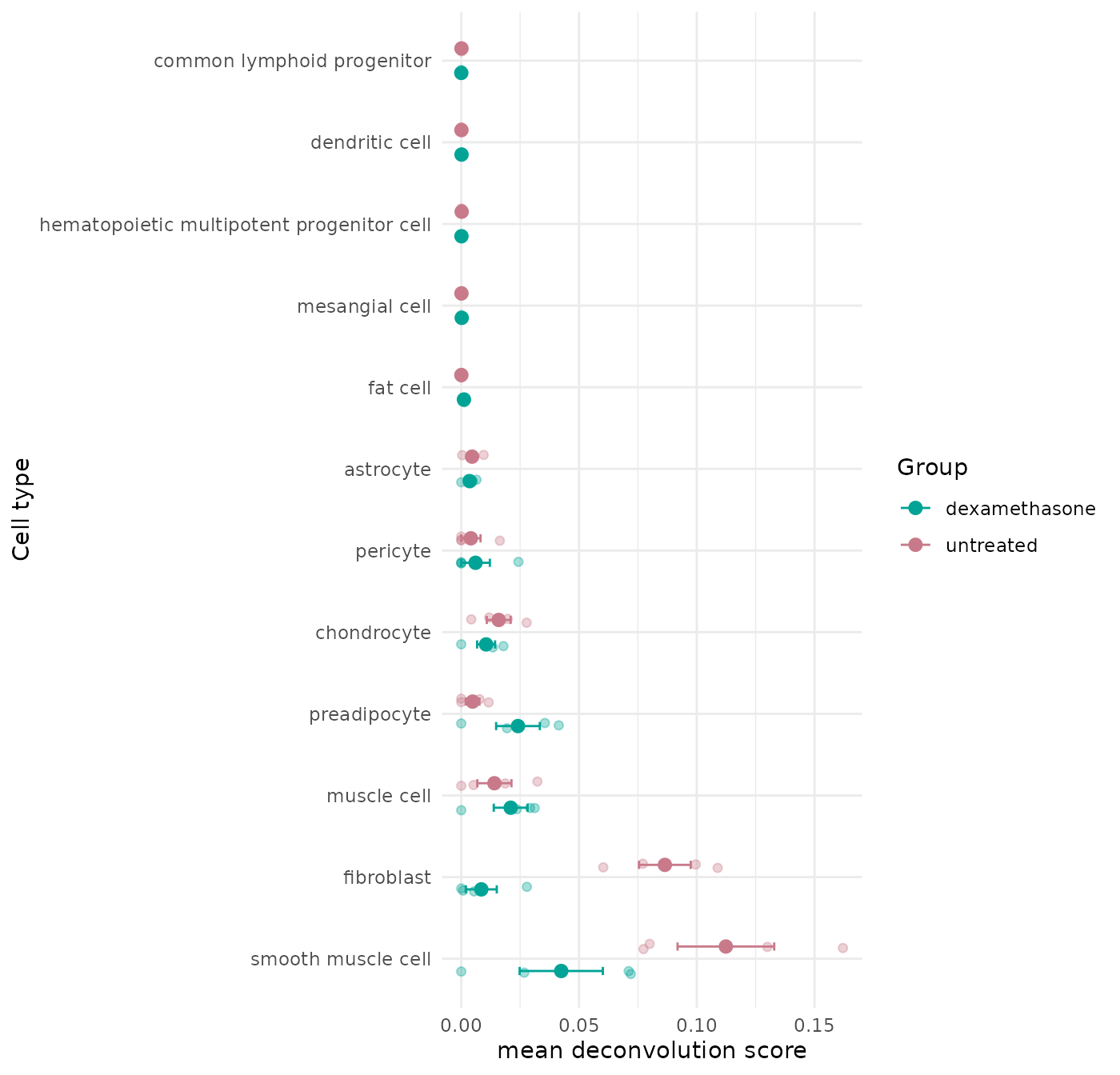
Group-Level Dot Plot
This plot summarizes deconvolution signal by treatment group while keeping sample-level points visible.
get_celltype_group_dotplot(
x = vista_deconv,
group_column = "cond_long",
top_n = 12,
summary_fun = "mean",
error = "se",
add_points = TRUE,
point_size = 2.5,
base_size = 11
)
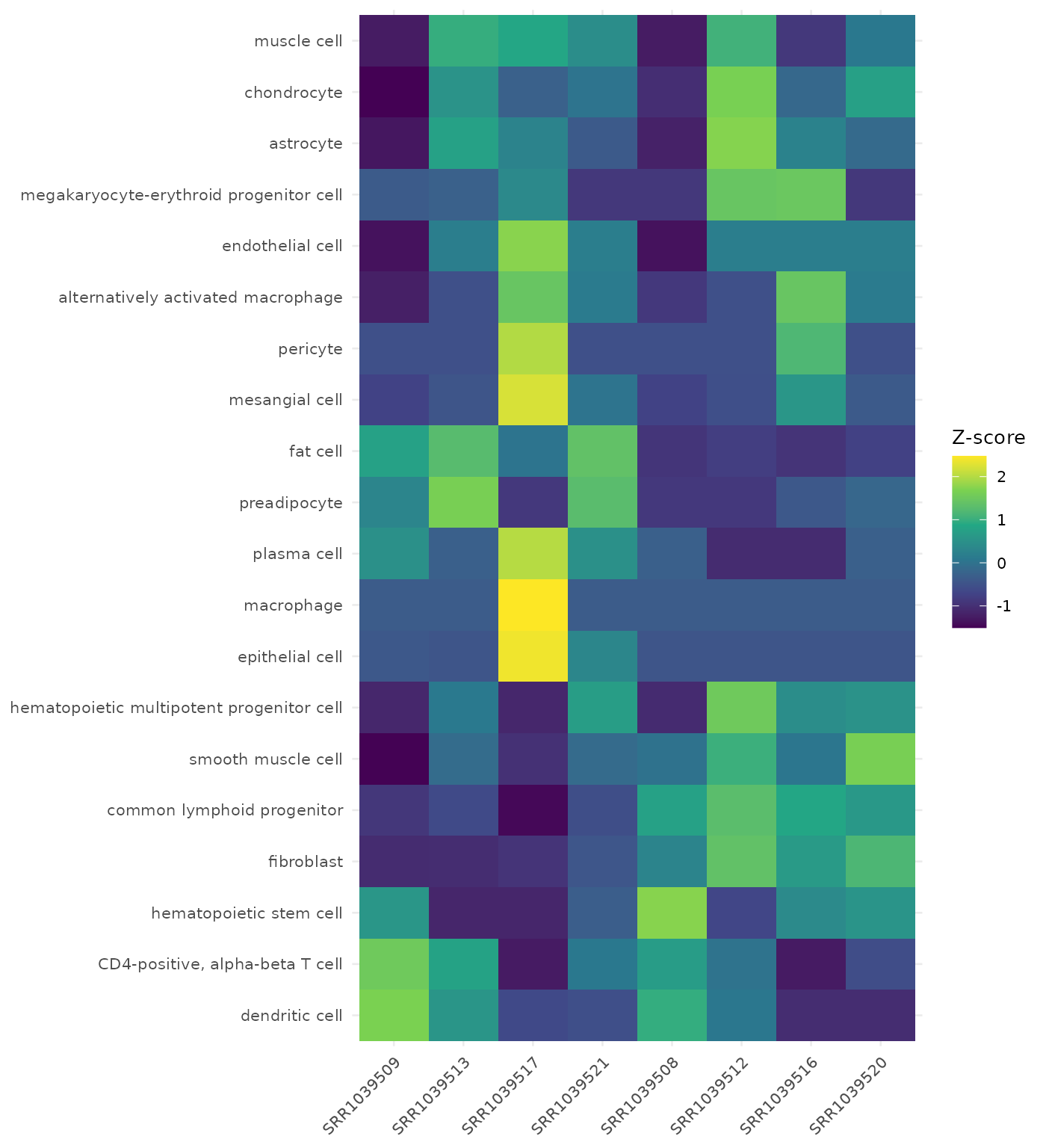
Cell-Type Heatmap
The heatmap is useful to inspect sample-level deconvolution structure and concordance within groups.
get_celltype_heatmap(
x = vista_deconv,
group_column = "cond_long",
top_n = 20,
transform = "zscore",
cluster_rows = TRUE,
cluster_columns = FALSE,
label = FALSE,
base_size = 10
)
Notes on Interpretation
- xCell2 outputs are typically enrichment-like abundance scores rather than absolute percentages.
- Use group-level differences and consistency across replicates as the primary interpretation signal.
- Treat results as hypothesis-generating; validate with orthogonal assays when possible.
Session Info
sessionInfo()
#> R version 4.5.3 (2026-03-11)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] org.Hs.eg.db_3.22.0 AnnotationDbi_1.72.0
#> [3] ggplot2_4.0.2 tidyr_1.3.2
#> [5] tibble_3.3.1 dplyr_1.2.0
#> [7] airway_1.30.0 SummarizedExperiment_1.40.0
#> [9] Biobase_2.70.0 GenomicRanges_1.62.1
#> [11] Seqinfo_1.0.0 IRanges_2.44.0
#> [13] S4Vectors_0.48.0 BiocGenerics_0.56.0
#> [15] generics_0.1.4 MatrixGenerics_1.22.0
#> [17] matrixStats_1.5.0 VISTA_0.99.4
#> [19] BiocStyle_2.38.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.5.3 filelock_1.0.3
#> [3] ggplotify_0.1.3 R.oo_1.27.1
#> [5] polyclip_1.10-7 graph_1.88.1
#> [7] minpack.lm_1.2-4 XML_3.99-0.23
#> [9] httr2_1.2.2 lifecycle_1.0.5
#> [11] rstatix_0.7.3 edgeR_4.8.2
#> [13] lattice_0.22-9 MASS_7.3-65
#> [15] backports_1.5.0 magrittr_2.0.4
#> [17] limma_3.66.0 sass_0.4.10
#> [19] rmarkdown_2.31 jquerylib_0.1.4
#> [21] yaml_2.3.12 otel_0.2.0
#> [23] ggtangle_0.1.1 cowplot_1.2.0
#> [25] DBI_1.3.0 RColorBrewer_1.1-3
#> [27] abind_1.4-8 quadprog_1.5-8
#> [29] purrr_1.2.1 R.utils_2.13.0
#> [31] msigdbr_26.1.0 pracma_2.4.6
#> [33] yulab.utils_0.2.4 tweenr_2.0.3
#> [35] rappdirs_0.3.4 gdtools_0.5.0
#> [37] enrichplot_1.30.5 ggrepel_0.9.8
#> [39] tidytree_0.4.7 annotate_1.88.0
#> [41] pkgdown_2.2.0 codetools_0.2-20
#> [43] DelayedArray_0.36.0 DOSE_4.4.0
#> [45] ggforce_0.5.0 tidyselect_1.2.1
#> [47] aplot_0.2.9 farver_2.1.2
#> [49] BiocFileCache_3.0.0 jsonlite_2.0.0
#> [51] Formula_1.2-5 systemfonts_1.3.2
#> [53] progress_1.2.3 tools_4.5.3
#> [55] ggnewscale_0.5.2 treeio_1.34.0
#> [57] xCell2_1.2.3 ragg_1.5.2
#> [59] Rcpp_1.1.1 glue_1.8.0
#> [61] SparseArray_1.10.10 xfun_0.57
#> [63] DESeq2_1.50.2 qvalue_2.42.0
#> [65] withr_3.0.2 BiocManager_1.30.27
#> [67] fastmap_1.2.0 GGally_2.4.0
#> [69] digest_0.6.39 R6_2.6.1
#> [71] gridGraphics_0.5-1 textshaping_1.0.5
#> [73] colorspace_2.1-2 GO.db_3.22.0
#> [75] RSQLite_2.4.6 R.methodsS3_1.8.2
#> [77] fontLiberation_0.1.0 data.table_1.18.2.1
#> [79] prettyunits_1.2.0 httr_1.4.8
#> [81] htmlwidgets_1.6.4 S4Arrays_1.10.1
#> [83] ontologyIndex_2.12 scatterpie_0.2.6
#> [85] ggstats_0.13.0 pkgconfig_2.0.3
#> [87] gtable_0.3.6 blob_1.3.0
#> [89] S7_0.2.1 SingleCellExperiment_1.32.0
#> [91] XVector_0.50.0 clusterProfiler_4.18.4
#> [93] htmltools_0.5.9 fontBitstreamVera_0.1.1
#> [95] carData_3.0-6 bookdown_0.46
#> [97] fgsea_1.36.2 zigg_0.0.2
#> [99] GSEABase_1.72.0 scales_1.4.0
#> [101] png_0.1-9 ggfun_0.2.0
#> [103] knitr_1.51 tzdb_0.5.0
#> [105] reshape2_1.4.5 nlme_3.1-168
#> [107] curl_7.0.0 cachem_1.1.0
#> [109] stringr_1.6.0 BiocVersion_3.22.0
#> [111] parallel_4.5.3 desc_1.4.3
#> [113] pillar_1.11.1 grid_4.5.3
#> [115] vctrs_0.7.2 ggpubr_0.6.3
#> [117] car_3.1-5 tidydr_0.0.6
#> [119] dbplyr_2.5.2 xtable_1.8-8
#> [121] cluster_2.1.8.2 singscore_1.30.0
#> [123] evaluate_1.0.5 readr_2.2.0
#> [125] cli_3.6.5 locfit_1.5-9.12
#> [127] compiler_4.5.3 rlang_1.1.7
#> [129] crayon_1.5.3 ggsignif_0.6.4
#> [131] labeling_0.4.3 plyr_1.8.9
#> [133] fs_2.0.1 ggiraph_0.9.6
#> [135] stringi_1.8.7 viridisLite_0.4.3
#> [137] BiocParallel_1.44.0 assertthat_0.2.1
#> [139] babelgene_22.9 Biostrings_2.78.0
#> [141] lazyeval_0.2.2 GOSemSim_2.36.0
#> [143] fontquiver_0.2.1 Matrix_1.7-4
#> [145] hms_1.1.4 patchwork_1.3.2
#> [147] bit64_4.6.0-1 KEGGREST_1.50.0
#> [149] statmod_1.5.1 AnnotationHub_4.0.0
#> [151] Rfast_2.1.5.2 igraph_2.2.2
#> [153] broom_1.0.12 memoise_2.0.1
#> [155] RcppParallel_5.1.11-2 bslib_0.10.0
#> [157] ggtree_4.0.5 fastmatch_1.1-8
#> [159] bit_4.6.0 ape_5.8-1
#> [161] gson_0.1.0